

Snippets of genetic material that have been linked to cancer also play a critical role in normal embryonic development in mice, according to a new paper from MIT cancer biologists.
The work, reported in the March 7 issue of Cell, shows that a family of microRNAs--short strands of genetic material--protect mouse cells during development and allow them to grow normally. But that protective role could backfire: The researchers theorize that when these microRNAs become overactive, they can help keep alive cancer cells that should otherwise die--providing another reason to target microRNAs as a treatment for cancer.
Discovered only a decade ago, microRNAs bind to messenger RNAs (mRNAs), preventing them from delivering protein assembly instructions, thereby inhibiting gene expression. The details of how microRNAs act are not yet fully understood.
"The scientific community is busy trying to understand what specific biological functions these microRNAs affect," said Andrea Ventura, lead author of the paper and postdoctoral associate in the Koch Institute for Integrative Cancer Research at MIT (formerly known as the Center for Cancer Research).
Ventura--who works in the laboratory of Tyler Jacks, director of the Koch Institute--and his colleagues studied the function of a family of microRNAs known as the miR-17~92 cluster.
Previous research has shown that the miR-17~92 cluster is overactive in some cancers, especially those of the lungs and B cells.
To better understand these microRNAs' role in cancer, the researchers decided to study their normal function. Knocking out microRNA genes and observing the effects can offer clues into how microRNA helps promote cancer when overexpressed.
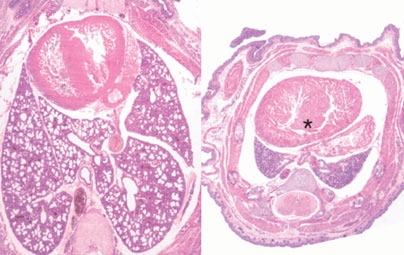
They found that when miR-17~92 was knocked out in mice, the animals died soon after birth, apparently because their lungs were too small. Also, their B cells, a type of immune cell, died in an early stage of cell development.
This suggests that miR-17~92 is critical to the normal development of lung cells and B cells. In B cells, these microRNAs are likely acting to promote cell survival by suppressing a gene that induces cell death, said Ventura.
"Understanding why these things are happening provides important insight into how microRNAs affect tumorigenesis," he said.
The researchers theorize that when miR-17~92 becomes overactive in cancer cells, it allows cells that should undergo programmed cell death to survive.
Blocking microRNAs that have become overactive holds promise as a potential cancer treatment. Research is now being done on molecules that prevent microRNAs from binding to their target mRNA.
More work needs to be done to make these inhibitors into stable and deliverable drugs, but Ventura said it's possible it could be done in the near future.
The exact genes targeted by miR-17~92 are not known, but one strong suspect is a gene called Bim, which promotes cell death. However, a single microRNA can have many targets, so it's likely there are other genes involved.
The researchers also studied the effects of knocking out two other microRNA clusters that are closely related to miR-17~92 but located elsewhere in the genome.
They found that if the other two microRNA clusters are knocked out but miR-17~92 remains intact, the mice develop normally. However, if miR-17~92 and one of these similar clusters are removed, the mice die before birth, suggesting there is some kind of synergistic effect between these microRNA families.
Other MIT authors of the paper are Amanda Young, graduate student in biology; Monte Winslow, postdoctoral fellow in the Center for Cancer Research (CCR); Laura Lintault, staff affiliate in the CCR; Alex Meissner, faculty member at the Broad Institute of MIT and Harvard; Jamie Newman, graduate student in biology; Denise Crowley, staff affiliate at the CCR; Rudolf Jaenisch, professor of biology and member of the Whitehead Institute for Biomedical Research; Phillip Sharp, MIT Institute Professor; and Jacks, who is also a professor of biology.
The research was funded by the National Institutes of Health and the National Cancer Institute.
A version of this article appeared in MIT Tech Talk on March 19, 2008 (download PDF).





